Our research effort is driven by the central question on how the three-dimensional structure of a biological macromolecule determines its biological function. We focus on the replication mechanism of herpesviruses and on exploring the involved macromolecules as potential antiviral therapy targets.
Current advances in computational protein design continuously push the boundaries of de novo protein design. We have implemented established protein packing algorithms into the in-house computer program MUMBO and have recently extended the scope of MUMBO to also redesign polynucleotides and protein-RNA/DNA interfaces. We are using these algorithms to design new ligand-binding sites into existing protein scaffolds and to design novel pairs of bacterial transcription regulators and RNA aptamers.
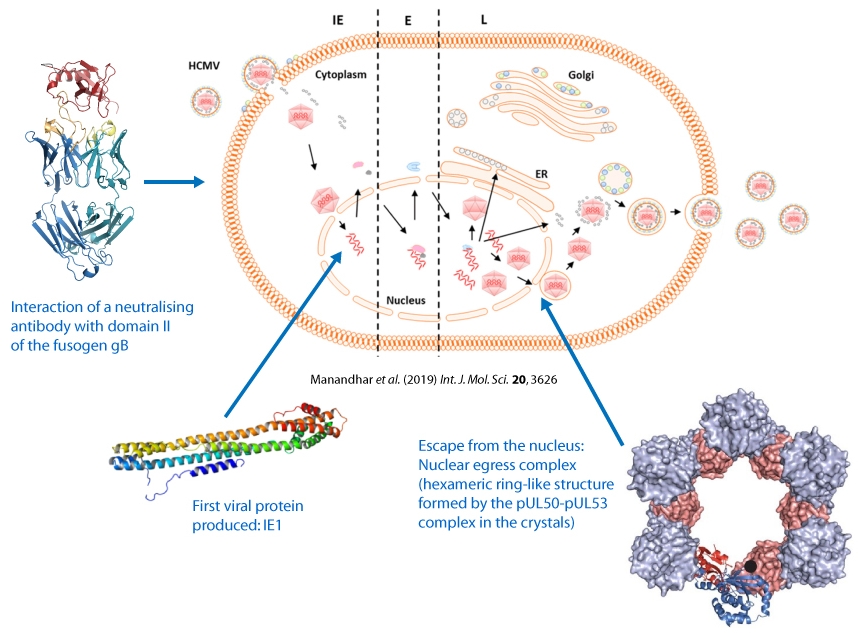
Through X-ray structure determinations of key viral proteins, we further our molecular understanding of specific steps of HCMV herpesviral replication.
Thus, we elucidated the mechanism by which neutralizing antibodies target the fusogenic protein gB presented on the surface of HCMV viral particles.
We determined the structure of the first viral protein that is produced by the virus in the host cell, i.e. Immediate Early Protein I (IE1).
We unraveled the architecture of the core nuclear egress complex (pUL50-pUL53 complex) that orchestrates the escape of virus particles from the nucleus.
Currently, we focus on the characterization of the molecular interactions that these proteins initiate with host cell proteins.
Collaboration partners:
Prof. Dr. M. Marschall, Virologie, Uniklinik Erlangen
Prof. Dr. J. Eichler, Pharmazeutische Chemie, FAU
Prof. Dr. T. Stamminger, Virologie, Universitätsklinikum Ulm
Spindler N., Diestel U., Stump J., Wiegers AK., Winkler T., Sticht H., Mach M., Muller Y.:
Structural Basis for the Recognition of Human Cytomegalovirus Glycoprotein B by a Neutralizing Human Antibody
In: PLoS Pathogens 10 (2014), S. e1004377
Scherer M., Klingl S., Sevvana M., Otto V., Schilling EM., Stump J., Müller R., Reuter N., Sticht H., Muller Y., Stamminger T.:
Crystal structure of cytomegalovirus IE1 protein reveals targeting of TRIM family member PML via coiled-coil interactions
In: PLoS Pathogens 10 (2014), S. e1004512
Walzer S., Egerer-Sieber C., Sticht H., Sevvana M., Hohl K., Milbradt J., Muller Y., Marschall M.:
Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions
In: Journal of Biological Chemistry 290 (2015), S. 27452-27458
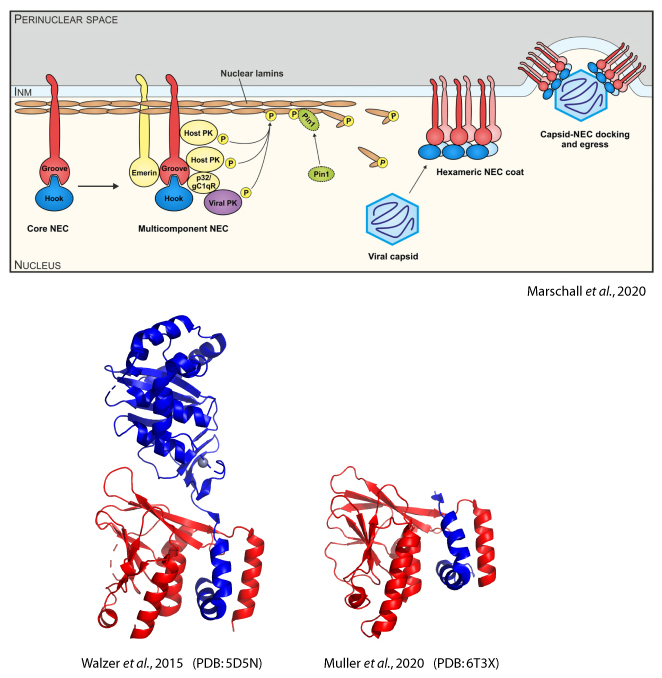
Herpesviruses are ubiquitous pathogens, some of which can cause life-threatening diseases (HCMV, KSHV). When herpesviral capsids have assembled in the host cell nucleus, all herpesviruses employ the conserved nuclear egress complex (NEC) to transport the capsids into the cytosol for further processing. NEC formation relies on the preceding formation of the heterodimeric core NEC, which is composed of the viral proteins pUL50 and pUL53 (HCMV notation).
A 29 amino acids long, hook-like extension of pUL53 binds into a groove within pUL50. Our group has previously solved crystal structures of both the pUL50-pUL53 complex (PDB: 5D5N) as well as pUL50 fused with the pUL53 hook (PDB: 6T3X), giving detailed insight into the interactions between pUL50 and pUL53 (Walzer et al., 2015, Muller et al., 2020). Besides HCMV, we are also interested in the NEC proteins from EBV, VZV and KSHV (Schweininger et al., 2022).
The hook-into-groove interaction makes the core NEC a promising target for novel antiherpesviral drugs. Using directed evolution and in silico methods, we aim to identify peptidic inhibitors of the hook-into-groove interaction that will serve as blueprints for future peptidomimetic or small molecule drugs.
References
Marschall, M., Häge, S., Conrad, M., Alkhashrom, S., Kicuntod, J., Schweininger, J., Kriegel, M., Lösing, J., Tillmanns, J., Neipel, F., Eichler, J., Muller, Y. A., & Sticht, H. (2020). Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses, 12(6), 683. https://doi.org/10.3390/v12060683
Walzer, S. A., Egerer-Sieber, C., Sticht, H., Sevvana, M., Hohl, K., Milbradt, J., Muller, Y. A., & Marschall, M. (2015). Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. The Journal of biological chemistry, 290(46), 27452–27458. https://doi.org/10.1074/jbc.C115.686527
Muller, Y. A., Häge, S., Alkhashrom, S., Höllriegl, T., Weigert, S., Dolles, S., Hof, K., Walzer, S. A., Egerer-Sieber, C., Conrad, M., Holst, S., Lösing, J., Sonntag, E., Sticht, H., Eichler, J., & Marschall, M. (2020). High-resolution crystal structures of two prototypical β- and γ-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. The Journal of biological chemistry, 295(10), 3189–3201. https://doi.org/10.1074/jbc.RA119.011546
Schweininger, J., Kriegel, M., Häge, S., Conrad, M., Alkhashrom, S., Lösing, J., Weiler, S., Tillmanns, J., Egerer-Sieber, C., Decker, A., Lenac Roviš, T., Eichler, J., Sticht, H., Marschall, M., & Muller, Y. A. (2022). The crystal structure of the varicella-zoster Orf24-Orf27 nuclear egress complex spotlights multiple determinants of herpesvirus subfamily specificity. The Journal of biological chemistry, 298(3), 101625. https://doi.org/10.1016/j.jbc.2022.101625
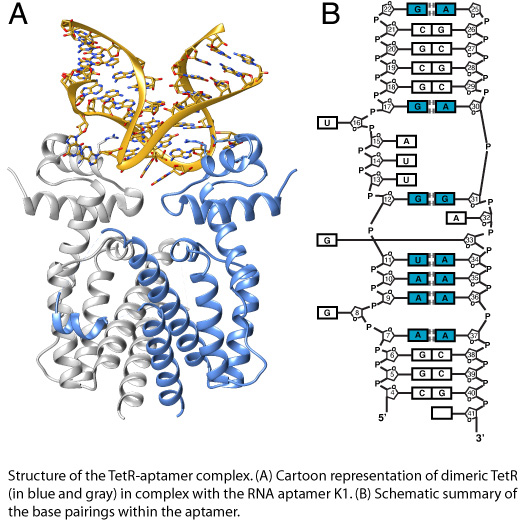
Grau F., Jaeger J., Groher F., Suess B., Muller Y.:
The complex formed between a synthetic RNA aptamer and the transcription repressor TetR is a structural and functional twin of the operator DNA-TetR regulator complex
In: Nucleic Acids Research 48 (2020), S. 3366-3378
Designer RNA molecules are increasingly explored as regulatory switches in synthetic biology. The TetR-binding RNA aptamer was selected by our collaboration partner for its ability to compete with operator DNA for binding to the bacterial repressor TetR. The fact that induction of TetR by tetracycline abolishes both RNA aptamer and operator DNA binding in TetR, enables numerous applications exploiting both the specificity of the RNA aptamer and the efficient gene repressor properties of TetR. Our recent crystal structure determination revealed the molecular determinants that are responsible for this functional behaviour.
Together with our collaboration partner, we currently seek to explore the synergistic potential of two approaches, i.e. computational design and molecular evolution, for the design of novel RNA aptamers directed against members of two different families of bacterial repressors. To achieve this goal, we have recently extended the scope of our computational design program MUMBO to also redesign polynucleotides and protein-RNA/DNA interfaces.
Collaboration partner:
Prof. Dr. B. Suess, Biologie, TU Darmstadt
Computer program MUMBO is available as a FORTRAN computer source from the software repository Gitlab (https://gitlab.com/group_muller/mumbo-software2.git) and/or from the authors upon request.
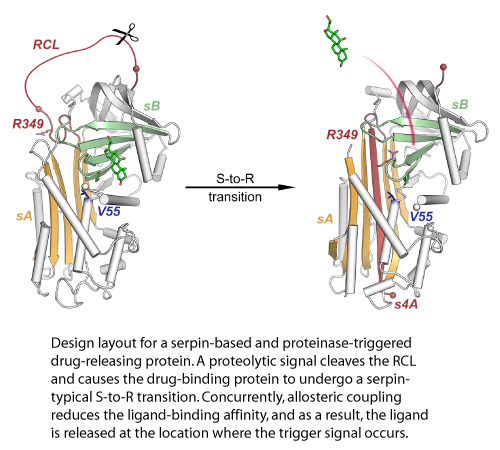
Schmidt K., Gardill B., Kern A., Kirchweger P., Börsch M., Muller Y.:
Design of an allosterically modulated doxycycline and doxorubicin drug-binding protein.
In: Proceedings of the National Academy of Sciences of the United States of America (2018)
The de-novo design of novel binding sites for ligands of our choice into existing protein scaffolds represents one of our key interests in computational protein design.
In a previous study, we were able to introduce doxycycline- and doxorubicin-binding sites into the serine proteinase inhibitor (serpin) family member α1-antichymotrypsin using the in-house computer program MUMBO. Further engineering allowed exploitation of the proteinase-triggered serpin-typical S-to-R transition to modulate the ligand affinities.
Taken together, this design strategy provides a blueprint for the design of novel binding proteins for the targeted delivery of therapeutic compounds.
Computer program MUMBO is available as a FORTRAN computer source from the software repository Gitlab (https://gitlab.com/group_muller/mumbo-software2.git) and/or from the authors upon request.